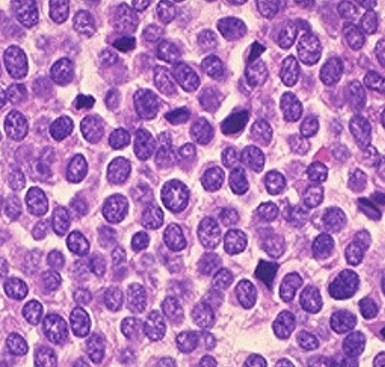
 Das Ewing-Sarkom ist ein seltener, bösartiger Tumor. Er tritt vorwiegend in der weißen Bevölkerung (kaukasisch) auf: Bei den unter 15-Jährigen sind ca. 3 von einer Million Menschen betroffen, bei den über 15-Jährigen sind es nur 2,4. In anderen Ethnien ist das Ewing-Sarkom noch seltener.
Das Ewing-Sarkom ist ein seltener, bösartiger Tumor. Er tritt vorwiegend in der weißen Bevölkerung (kaukasisch) auf: Bei den unter 15-Jährigen sind ca. 3 von einer Million Menschen betroffen, bei den über 15-Jährigen sind es nur 2,4. In anderen Ethnien ist das Ewing-Sarkom noch seltener.
Die Erkrankung tritt vor allem bei Kindern, Jugendlichen und jungen Erwachsenen auf. Dabei sind Jungen und Männer etwas häufiger betroffen als Mädchen und Frauen (1,5:1).
Grundsätzlich kann das Ewing-Sarkom überall im Körper auftreten: 85 % gehen vom Knochen, 15 % vom Weichteilgewebe aus. Am häufigsten entstehen Ewing-Sarkome in den Beckenknochen, gefolgt von den Oberschenkel- und Unterschenkelknochen. Das Ewing-Sarkom des Weichteilgewebes findet man eher bei Erwachsenen über 30 Jahren.
Die Ursache für die Entstehung des Tumors ist unbekannt. Weder eine familiäre Veranlagung noch der Kontakt mit Chemikalien, Bestrahlung oder andere Umweltfaktoren scheinen eine Rolle zu spielen.
Allerdings gibt es bestimmte Genveränderungen und -umlagerungen, die offenbar typisch für das Ewing-Sarkom sind.
Diagnose
Besteht der Verdacht auf ein Sarkom, dann wird in einem ersten Schritt die Krankengeschichte erfasst (Anamnese) und eine sorgfältige körperliche Untersuchung durchgeführt. Sollte eine Schwellung vorliegen, kommen zusätzlich bildgebende Verfahren zur Unterstützung der Diagnose und zum Staging (Einstufung der Bösartigkeit) zum Einsatz.
Es gibt verschiedene Verfahren, die zur Diagnose des Ewing-Sarkoms zum Einsatz kommen können:
- Röntgenaufnahmen des betroffenen Bereichs
- Magnetresonanz- bzw. Kernspintomografie (MRT) zur Bestätigung der Diagnose, besonders dann, wenn der Tumor außerhalb des Knochens entstanden ist
- Computertomografie (CT): Sie misst die Ausdehnung und Größe des Tumors und soll feststellen, ob Metastasen, vor allem in der Lunge (Thorax-CT), vorhanden sind
- Ein Knochenszintigraphie kann möglicherweise vorhandene Krebsherde im Knochen aufzeigen
- Die Positronen-Emissions-Tomografie (PET) oder PET-CT bzw. PE-MRT kommt derzeit nur in bestimmten Fällen zum Einsatz und gehört nicht zu den Standard-Diagnoseverfahren beim Ewing-Sarkom.
 Um die Diagnose zu sichern bzw. endgültig festzulegen, ist es notwendig, eine Gewebeprobe zu entnehmen (Biopsie). Diese sollte unbedingt von Sarkom-Spezialisten durchgeführt werden: Sie sind in der Lage, die bestmögliche Einstichstelle zu wählen und damit die Gefahr von Komplikationen zu minimieren.
Um die Diagnose zu sichern bzw. endgültig festzulegen, ist es notwendig, eine Gewebeprobe zu entnehmen (Biopsie). Diese sollte unbedingt von Sarkom-Spezialisten durchgeführt werden: Sie sind in der Lage, die bestmögliche Einstichstelle zu wählen und damit die Gefahr von Komplikationen zu minimieren.
Darüber hinaus ist in der Regel auch eine Knochenmarkbiopsie erforderlich. Dabei wird etwas Knochenmark und ein winziges Knochenstück durch eine Hohlnadel entnommen, die in den Hüftknochen eingeführt wird. Diese Proben werden unter dem Mikroskop untersucht, um festzustellen, ob sich die Krankheit bereits ausgebreitet hat.
|
Besonders beim Ewing-Sarkom: Das EWSR1-FLI1 Fusionsgen Bei vielen Krebsarten, die bei Erwachsenen vorkommen, wird oft eine große Anzahl an Gen-Mutationen nachgewiesen. Bei Kindern und/oder jungen Erwachsenen hingegen liegen bei den meisten bösartigen Erkrankungen einschließlich des Ewing-Sarkoms nur wenige Mutationen vor. Ewing-Sarkome weisen eine Mutation des EWSR1-Gens auf, das sich mit einer Reihe anderer Gene verbinden kann. Bei über 90 % der Patienten wird diese Mutation nachgewiesen. Allerding kann der Krankheitsverlauf sowie das Ansprechen auf eine Therapie sehr unterschiedlich sein. Aktuell versuchen Forscher, mehr über die Biologie des Ewing-Sarkoms herauszufinden, um dann zielgerichtete (targeted) bzw. personalisierte Therapien entwickeln zu können. |
Behandlung
Die Behandlung des Ewing-Sarkoms sollte in einem Sarkomzentrum erfolgen, ggf. im Rahmen einer klinischen Studie. Hier erhalten die Patienten eine Therapie, die ihrem Krankheitsstadium, aber auch weiteren Faktoren wie dem Ansprechen auf eine systemische Therapie und/oder der Tumorgröße angepasst wird.
 Ein fachübergreifendes Team aus (pädiatrischen) Onkologen, Radiologen, Chirurgen, Pädiater, Pathologen u.s.w. schaut sich die persönliche Krankengeschichte, das vorhandene Bildmaterial und die Ergebnisse der Gewebeuntersuchung an und bespricht die Behandlungsoptionen. Ziel ist die Entwicklung Behandlungsplans für den einzelnen Patienten. Dieser umfasst normalerweise eine Operation und/oder die Strahlentherapie zur lokalen Kontrolle des Tumors sowie eine Chemotherapie als systemische (den ganzen Körper betreffend) Maßnahme.
Ein fachübergreifendes Team aus (pädiatrischen) Onkologen, Radiologen, Chirurgen, Pädiater, Pathologen u.s.w. schaut sich die persönliche Krankengeschichte, das vorhandene Bildmaterial und die Ergebnisse der Gewebeuntersuchung an und bespricht die Behandlungsoptionen. Ziel ist die Entwicklung Behandlungsplans für den einzelnen Patienten. Dieser umfasst normalerweise eine Operation und/oder die Strahlentherapie zur lokalen Kontrolle des Tumors sowie eine Chemotherapie als systemische (den ganzen Körper betreffend) Maßnahme.
Eine Chemotherapie wird oft vor einer Operation (neoadjuvante Therapie) oder der Bestrahlung eingesetzt, um den Tumor zu verkleinern und um ggf. Tumorzellen abzutöten, die sich bereits vom Primärtumor (Ursprungstumor) gelöst haben. Auch nach einem chirurgischen Eingriff kann Chemotherapie verabreicht werden, um zu verhindern, dass der Tumor zurückkehrt (adjuvante Therapie).
Der chirurgische Eingriff, also die Operation, zielt darauf ab, den Tumor (oder die Reste, die nach Chemotherapie oder Bestrahlung noch vorhanden sind) möglichst vollständig zu entfernen. Wenn Knochensubstanz und/oder andere Gewebe durch einen solchen Eingriff verlorengehen, werden sie entweder durch körpereigenes bzw. gespendetes Gewebe oder ein Implantat ersetzt.
Eine Bestrahlung kommt dann zum Einsatz, wenn der Tumor durch eine Operation nicht entfernt werden kann, oder wenn eine Operation wichtige Körperfunktionen schwer beeinträchtigen würde. Nach einer Operation kann die Strahlentherapie genutzt werden, um mögliche Tumorreste oder Metastasen zu behandeln. Das Ziel ist es, zu verhindern, dass der Tumor zurückkehrt.
Für die meisten Ewing-Patienten erfolgt eine Behandlung in mehreren Schritten:
- Chemotherapie zur Verkleinerung des Tumors
- Operation zur Tumorentfernung
- Bestrahlung zum Abtöten verbliebener Tumorzellen oder Metastasen
- Chemotherapie zur Abtötung von Krebszellen, die sich ggf. vom Primärtumor gelöst haben und im Körper vorhanden sind
Nachsorge
 Konnte der Tumor komplett entfernt werden, sollten regelmäßige Nachuntersuchungen erfolgen, um so früh wie möglich zu erkennen, wenn ein Tumor wieder auftritt.
Konnte der Tumor komplett entfernt werden, sollten regelmäßige Nachuntersuchungen erfolgen, um so früh wie möglich zu erkennen, wenn ein Tumor wieder auftritt.
Wie oft und welche Nachuntersuchungen notwendig sind, hängt von mehreren Faktoren ab: Die Gewebeart, das Stadium der Krebserkrankung bei der Diagnose und die bisherigen Therapien werden hierzu in Betrachtung gezogen. Auch Blutuntersuchungen oder erneute Bildaufnahmen können nötig werden.
Spätfolgen
 Einige Folgen von Chemotherapie oder Bestrahlung können erst Monate oder sogar Jahre nach Abschluss der Behandlung auftreten. Man spricht von Spätfolgen. Sie können fast überall im Körper entstehen. Dabei kann es sich um Herz- oder Lungenprobleme handeln, um eine zweite Krebserkrankung, aber auch um Angstgefühle, Depression und Lernschwierigkeiten. Bei Kindern kann es zu Auffälligkeiten in der Entwicklung oder bei der Fruchtbarkeit kommen, sowie zu einer Verzögerung des Wachstums.
Einige Folgen von Chemotherapie oder Bestrahlung können erst Monate oder sogar Jahre nach Abschluss der Behandlung auftreten. Man spricht von Spätfolgen. Sie können fast überall im Körper entstehen. Dabei kann es sich um Herz- oder Lungenprobleme handeln, um eine zweite Krebserkrankung, aber auch um Angstgefühle, Depression und Lernschwierigkeiten. Bei Kindern kann es zu Auffälligkeiten in der Entwicklung oder bei der Fruchtbarkeit kommen, sowie zu einer Verzögerung des Wachstums.
Ewing-Patienten, die sich einer solchen Therapie unterzogen haben, haben ein erhöhtes Risiko für solche Spätfolgen, verglichen mit der Allgemeinbevölkerung. Sie sollten daher über einen längeren Zeitrahmen hinweg nachbeobachtet werden.
 Neue Behandlungsmöglichkeiten für das Ewing-Sarkom werden derzeit im Rahmen klinischer Studien untersucht. Diese schließen neue zielgerichtete Therapien wie monoklonale Antikörper, Immuntherapie oder andere neue Medikamente ein.
Neue Behandlungsmöglichkeiten für das Ewing-Sarkom werden derzeit im Rahmen klinischer Studien untersucht. Diese schließen neue zielgerichtete Therapien wie monoklonale Antikörper, Immuntherapie oder andere neue Medikamente ein.